
Measuring cancer evolution from the genome
Measuring cancer evolution from the genome
Graham, T. A*., & Sottoriva, A*. (2016). Measuring cancer evolution from the genome. The Journal of Pathology, 241(2), 183–191. doi:10.1002/path.4821
- Evolution and Cancer Laboratory, Barts Cancer Institute, Charterhouse Square, Queen Mary University of London, EC1M 6BQ, UK
- Cancer Evolutionary Genomics and Modelling Laboratory, Centre for Evolution and Cancer, The Institute of Cancer Research, Sutton, SM2 5NG, UK
* Correspondence to: Trevor A Graham, Evolution and Cancer Laboratory, Barts Cancer Institute, Charterhouse Square, Queen Mary University of London, EC1M 6BQ, UK. Phone: +44 20 7882 6231. Email: t.graham@qmul.ac.uk, and Andrea Sottoriva, Cancer Evolutionary Genomics and Modelling Laboratory, Centre for Evolution and Cancer, The Institute of Cancer Research, Sutton, SM2 5NG, UK. Phone: +44 20 8722 4072. Email: Andrea.Sottoriva@icr.ac.uk
Conflict of interest statement: The authors have no conflicts of interest to declare. No writing assistance was used in the preparation of this manuscript.
Abstract
The temporal dynamics of cancer evolution remain elusive because it is impractical to longitudinally observe cancers unperturbed by treatment. Consequently, our knowledge of how cancers grow largely derives from inferences made from a single point in time – the end point in the cancer’s evolution when it is removed from the body and studied in the lab. Fortuitously however, the cancer genome, by virtue of on-going mutations that uniquely mark clonal lineages within the tumour, provides a rich yet surreptitious record of cancer development. In this review, we describe how a cancer’s genome can be analysed to reveal the temporal history of mutation and selection, and discuss why both selective and neutral evolution feature prominently in carcinogenesis. We argue that selection in cancer can only be properly studied once we have a handle on what the absence of selection looks like. We review the data describing punctuated evolution in cancer, and reason that punctuated phenotype evolution is consistent with both gradual and punctuated genome evolution. We conclude that to map and predict evolutionary trajectories during carcinogenesis it is critical to better understand the relationship between genotype change and phenotype change.
Keywords
Clonal evolution of cancer, neutral evolution, selection, punctuated equilibrium, gradualism, saltation, hopeful monsters, subclones, intra-tumour heterogeneity, next generation sequencing.
Introduction
How do cancers grow? This basic question continues to be difficult to answer for the obvious reason that longitudinal observation of tumour growth is nearly always impractical, both in human and also in model systems. Consequently, our understanding of tumour formation relies on historical inference based on the composition of excised tumours. In other words, our understanding of the temporal process of tumour evolution is largely derived from data collected at a single time point: the time point at the end of the process when the tumour ends up on the specimen table. But this state of affairs is not as sorry as it may sound, as fortunately the tumour genome (or more accurately genomes of all the cells in the tumour) provides a surreptitious yet rich record of a tumour’s growth.
Each time a cell divides, errors during DNA replication mean that new mutations are introduced into the genomes of the daughter cells[1-3]. Epigenetic marks (e.g. DNA methylation) are also copied with limited fidelity[4]. Larger-scale chromosomal or part- chromosome losses or amplifications (somatic copy number alteration – SCNA) and other structural rearrangements also occur at an appreciable frequency in many cancers[5, 6]. It is these naturally occurring (epi)genetic alterations that record the ancestry of the cells in the tumour, and because tumours are clonally derived all of the cells in the tumour will carry the mutations in the first cancer cell, whereas later arising subclones are identifiable by their sharing of a particular unique set of variants, and so the order of clone development can be inferred by comparing the sets of mutations present in different cells of the tumour. The logic of this kind of analysis is at the heart of phylogenetics methods as applied to cancer[7]. Moreover, if a particular type of mutation accrues at a constant rate (e.g. the same number of new mutations are introduced in each cell division; this appears to be the case for C>T transitions within specific 3-base pair motifs for example[1]), then counting the number of mutations of the type that are unique to a particular lineage gives an estimate of the relative time that the lineage arose. A constant mutation rate is referred to as a molecular clock, and if the rate at which the molecular clock ‘ticks’ is known, then the absolute time of events (where time is measured in cell divisions elapsed) can also be determined[8]. These methods have been applied to a wide variety of cancers and revealed new insight into the order and timing of mutation accumulation (for some examples see refs [9-25]).
But mutation is not the only force shaping the cancer genome: evolutionary selection also plays a critical role. Selection describes that the situation where one group of cells within the tumour is evolutionary ‘favoured’ over another, such that the favoured cells have more offspring than the not-favoured cells. The favour is a result of the cell evolving a new phenotypic trait that gives it an advantage in the current microenvironment (context) of the tumour; the trait is referred to as adaptive. For example, a cell with a low metabolic demand might grow faster than a cell with a high metabolic need when both cells are together in a nutrient-poor microenvironment. The result of selection is that any mutation in the selected (favoured) population become more common in the tumour population as a whole, whereas negatively selected clones (not-favoured) become relatively less common.
Consequently, selection plays a central role in shaping the frequency distribution of mutations within a tumour.
To understand how a tumour has grown from its genome, we therefore need to understand both mutation and selection, and critically how these two processes together shape the pattern and frequency of mutations in the genome. Mutation and selection are deeply intertwined, since a new mutation may produce a new adaptive trait and therefore drive selection, and conversely a new microenvironmental selective pressure (such as targeted therapy, for instance) may mean that a pre-existing mutation becomes adaptive and so increases in frequency[26]. In general, mutation is considered a random process; any mutation may occur at any time with some (low and/or fluctuating) probability, whereas selection is non-random; only particular mutations are adaptive in a given context[27, 28]. For example, loss of normal function of the APC gene provides a clear selective advantage to cells in the intestine[29-31], but not, say, in the lung, even though presumably APC mutation occurs at a comparable rate in both tissues. Thus, the frequency at which particular mutations are observed across tumour is a function both of the rate at which the mutations occur, and also of the likelihood the mutation has being adaptive and driving a clonal expansion to a detectable level.
Tissue architecture –which we can broadly think of as the ‘mechanical microenvironment’ – provides additional selective constraints on tumour evolution. Many epithelia have glandular architecture (eg the crypts in the colon, and ducts in the prostate and breast), and
it is the abnormal growth of these glands (rather than the cells within them per se) that underlies neoplastic growth. Thus, cancer development requires evolution to at multiple levels in epithelial tissues[32]: in the example of the colon, first a mutated cell must repopulate the crypt, and then the mutant crypt itself must divide to form a glandular adenoma[33]. Computational modelling suggests that these tissue architectures have (at least in part) evolved to suppress clonal evolution[34]. Tissue architecture means that in solid tumours clonal expansions are spatially delineated, and so the indicators of mutation and selection within the genome are likely to show intra-tumour heterogeneity.
It is clear that cancer formation requires the acquisition of a number of key driver alterations (mutations and epigenetic changes in cancer cells) [35, 36]. The precise number of drivers per cancer is uncertain – and indeed given the inherently contextual nature of selection a comprehensive list of cancer-specific drivers is unlikely to exist in reality. It serves our purposes here to think of driver mutations as mutations that are positively selected in their (changing) microenvironmental context within the tumour. A central question in tumour evolution is: what is the temporal pattern of driver alteration acquisition? The competing theories are gradualism and punctuation (see Table 1 for definition of terms). The gradualist theory proposes that cancer evolution happens via a steady accumulation of driver mutations and a concomitant steady series of selective clonal outgrowths, whereas the punctuated theory proposes that the evolution of cancer occurs in fits and starts.
In this review, we address how mutation and selection together shape the cancer genome, with particular reference to the manifestations of graduated and punctuated evolution.
Detecting selection and neutrality from the cancer genome
Clonal selection, whatever the biological mechanism driving it, ultimately results in the relative outgrowth of the selected clone within the tumour. The clonal outgrowth appears in the cancer’s genome as an ‘over-representation’ of the mutations in the selected clone, as compared to the ‘null’ case where the genome evolved in the absence of selection (Figure 1). In principle then, detecting selection just requires spotting the characteristic ‘clonal outgrowths’, and many different bioinformatics tools have been developed to spot the ‘clusters’ of mutations at similar frequency in tumour next generation sequencing data that are characteristic of these outgrowths[22, 37-39]. It is important to note here that the evolutionary dynamics of the selected clone are largely revealed by the passenger mutations in that clone, not the drivers themselves: as the selected clone grows out, all of the many passenger mutations in the clone are carried along to higher frequency, making the selected clone visible against the milieu of unselected mutations in the tumour. Therefore, both driver and passenger mutations in the clone are affected by selection, but passenger mutations are generally more informative as they are more numerous[40]. This is just because, as evolution is a blind force, for every ‘successful’ driver mutation, many ‘unsuccessful’ mutations have occurred in a genome as large as the human one.
Moreover, this means that clonal selection is always visible in the frequency distribution of mutations in cancer irrespective of the biological mechanism that provides the selective advantage. For example, suppose that rather than the acquisition of a new driver mutation, a clone becomes a selective advantage because of a sudden change in microenvironmental context (such as a new non-cell autonomous interaction within the tumour[41]); even though the clone’s advantage is cell-extrinsically driven, its passenger mutations will still become overrepresented.
However, we argue that to be able to reliably spot clonal outgrowths in a cancer’s genome, we first require an understanding of what the ‘null case’ – evolution in the absence of clonal selection – looks like. The absence of clonal selection is referred to as neutral evolution, and by definition neutral evolution (in a growing population like a tumour) is the case when all cells grow at the same rate as one another. The definition of neutral evolution also encompasses stochastic drift. In a drifting population, all cells growth at the same average rate, but at any single point in time any one lineage might, because of random effects, grow or shrink slightly faster than another. We note that if a ‘lucky’ clone happens to drift to proportionally high frequency in a neutrally evolving asexual population like cancer, it could appear indistinguishable from a selected clone.
Mathematical modelling (or perhaps more accurately put: population genetics theory) provides a formal description of the frequency of subclonal mutations within a neutrally growing tumour[42-44]. Under neutrality the cumulative number of mutations at frequency f follows a ‘1/f’ distribution: this means that the number of mutations at a particular frequency in the tumour will double each time the frequency halves, or more loosely, when a tumour is growing neutrally there will be ever more mutations at ever lower frequencies. This mathematical result can be understood intuitively: because a small number of new mutations are expected to accrue each time a cell divides, then as the tumour population increases in size, more and more new mutations are accrued by the population as a whole, and the ‘1/f’ distribution is reached because precisely twice as many new mutations are expected each time the population doubles in size. To test for selection in a growing cancer, it suffices therefore to ask whether or not the distribution of mutation frequencies observed in the cancer (as measured by next generation sequencing) follows a 1/f distribution: in the case where they do not, we can reject the null hypothesis of neutrality in favour of recent selection.
This ‘1/f test’ must be applied with caution. Limited depth sequencing can blur the signal from the evolutionary dynamics (the signal of both selection and neutrality alike) [42]. It is conceivable that a particular ‘just-right’ combination of subclones could produce a VAF distribution that masquerades as a 1/f distribution (if the selected subclones happened to reach a particular set of sizes and the ‘noise’ in the sequencing data blurred their passenger mutation VAFs appropriately). While the allele frequencies alone cannot discount this possibility, nevertheless neutral evolution provides a much more parsimonious explanation of a 1/f –like VAF distribution. Moreover, we note that the 1/f test provides an objective indication of the presence of absence of subclonal selection, which does not require prior knowledge of the identity of subclonal drivers.
The ratio of non-synonymous (NS) mutations (that are likely to alter fitness by changing protein structure and function) to synonymous (S) mutations (that are likely neutral) at a particular locus is another popular test for selection[45, 46]. Typically, the NS/S ratio is normalised by the number of possible NS and S mutations that can occur at the locus of interest (the normalised ratio is referred to as dN/dS), and then deviations in the normalised ratio above 1 indicate positive selection (more NS mutations than expected by chance) whereas deviations below 1 indicate negative selection (fewer NS mutations than expected by chance). Applying the dN/dS ratio to cancer is complicated by the differential and evolving mutation rates of 3-basepair motifs[47], that can potentially skew the dN/dS values, but nevertheless, corrected dN/dS ratios within large cohorts of tumours has revealed evidence of positive selection in cancer on particular gene sets, such as the kinases[48]. We note that applying dN/dS to detect subclonal selection within a tumour is extremely challenging, because if the selection is caused by a single base-pair change (e.g. the common KRAS c.35G>T mutation) then the signal from this locus will be ‘drowned out’ by all the other passenger mutations within the clone, and applying dN/dS on a gene-by- gene basis in individual tumours is not possible because of the relatively low numbers of detected somatic mutations in any individual cancer.
How often does selection occur?
We recently looked for evidence of clonal selection across cancer types using the ‘1/f’ test described above. Remarkably, our analysis showed that in approximately 30% of cancers of 14 different solid cancer types we were unable to reject the null hypothesis of neutral evolution[42]. In cancer model systems, neutral drift of tumour cells is also observed[49]. Therefore, the signature of selection appears to be somewhat rarer than we might naively have expected from a gradualistic evolutionary perspective.
How often should we expect to see selection manifested in the cancer genome?
First, we only expect to see a clonal outgrowth if the clone is ‘caught in the act’ of growing out – e.g. if the clone had already expanded to at least a minimal detectable size at the time the tumour was sampled, but before it has expanded to repopulate the entire tumour[50]. The latter point is because once a selected clone has taken over the whole tumour then all the cells within the clone are the same as one another – and so the population then again evolves neutrally. The duration of time where a clone can be ‘caught in the act’ of expanding is determined by the selective advantage of the clone (relative to the residual ‘host’ cells in the tumour): fitter clones will grow out quicker. Unfortunately, empirical measurements of selective advantages of clones in growing tumours are lacking, so our expectations of the likelihood of detecting a selected clone mid-expansion are largely guesswork. Within the intestinal crypt (a constant population size) empirical measurement of the selective advantages of the tumour suppressor gene APC and the proto-oncogene KRAS reveal almost two-fold increases in the probability of stem cell replacement[29]. If tumour subclones experienced similarly large selective advantages, we might expect to only rarely see partially-expanded clones. Intriguingly though, abstract mathematical modelling of mutation accumulation in growing tumours suggests that very low selective advantages for
new driver mutations (of the order of less than 1%) lead to reasonable waiting times to cancer (in the models, cancer is defined by a subclone having accumulated a critical driver mutation burden) [51, 52]. In addition, our own computational modelling shows that even sizeable selective advantages produce only slight changes in clone frequency in a growing population, and this result is exacerbated when a new clone is formed in an already large tumour[14]. Together, these results would predict partially-expanded subclones would be commonplace, if they were initiated at sufficiently high rate. Clearly, empirical measurement of the differential fitness of tumour subclones is required.
Second, the likelihood of seeing selection is also determined by the rate at which new selected clones are generated, either by clone-intrinsic mutation or the creation of a favourable microenvironment. This rate is directly related to the number of potential driver alterations a clone can acquire: if there are many potential drivers then new driver mutations are likely to occur frequently. Interestingly, genome sequencing studies on large cohorts (such as the Cancer Genome Atlas – http://cancergenome.nih.gov/) consistently reveal fairly short lists of recurrently-mutated genes in each cancer type: for example in a cohort of 276 colorectal cancers only 24 genes were mutated at significantly greater than background frequency[53]. These studies suggest that the number of drivers may actually be quite limited, and hence neutral dynamics may be relatively common in cancers because of the low rate of driver mutation accrual.
Third, our ability to detect selection is, of course, limited by the resolution of our tools to look for it. The current standard of moderate depth exome sequencing is 100X coverage, facilitating reasonably reliable detection of mutations at about 5% frequency. This means that low selective advantages that cause only slight changes in clone frequency are largely indistinguishable from the background neutral evolution. The ‘mini-driver’ hypothesis, which postulates that there are many mutations each causing small fitness effects in cancers[54], would clearly be challenging to confirm or refute from moderate depth sequencing data. Moreover, as a tumour grows, newly generated clones form ever lower proportions of the tumour cell population and so detecting them because ever more challenging as the tumour becomes larger: thus the ~5% sensitivity of sequencing provides a window to detect only those clones that form very early in a cancer’s grow, or those which rapidly (e.g. within a small proportion of the lifetime of the cancer) grow to a detectable
Evolutionary dynamics and tumour progression
Manifest on-going selection in cancers appears to be associated with a worse prognosis because across cancer types, tumours with three or more large clones have a worse prognosis than tumours with fewer clones[55, 56], and putative subclonal driver mutations are also associated with a worse prognosis[55].
However, neutral evolution has a potential ‘dark side’ for prognosis, by virtue of it allowing huge variation to be generated and persist in a tumour. Whilst by definition the diversity in a neutrally evolving tumour is non-adaptive to the current microenvironment, if the microenvironment were to change – through the application of therapy for instance – then variants within this reservoir of pre-existing variation could suddenly become adaptive.
Thus neutrally evolving tumours may be particularly prone to develop therapy resistance. The relationship between neutral and selective evolutionary dynamics and tumour progression should be the focus of future work.
Our recent analysis of the evolution of colorectal cancer led us to put forward the “Big Bang” model of cancer growth, whereby the tumour mass grows as a single clonal expansion wherein differential clonal selection within the tumour has little influence on the subclonal composition of the tumour, and instead clonal mosaicism is determined largely by the time of clone generation[14]. Since the clonal composition of a Big Bang tumour is determined simply by which clones were generated at the beginning of cancer growth, we speculated that a tumour’s prognosis is similarly predetermined. In other words, in the absence of clonal selection, the phenotype of the ‘first’ cancer cells should determine the cancer’s behaviour thereafter. Consequently, we speculate that reading these ‘initial phenotypes’ in the grown cancer may be prognostic, for example by looking for the degree of clonal mixing as a read-out of cell migration ability, or the degree to which a clone coexists in multiple different microenvironments as a readout of plasticity.
Punctuated evolution
In the evolutionary biology literature, punctuated evolution is (loosely) defined as an
apparently abrupt change in phenotype, and was originally suggested by Eldredge and Gould to explain the large morphological differences between species that appeared to occur without the presence of intermediate morphotypes in the fossil record[57]. The original descriptions of punctuated evolution put forward that an ancestral species became subdivided into (spatially) isolated distinct niches where each subpopulation independently (and gradually) evolved until the point that one of those sub-species – by now grossly altered compared to the ancestor – was able to escape the niche and expanded its population significantly (Figure 2A). Because the isolating niche was small, the intermediate forms were lost to the fossil record, and only the widespread ancestral and then the new grossly-altered populations were captured. The result was an apparently punctuated evolution of species – interspersed by long periods of time where apparently no ‘important evolution’ happened. This pattern of events was described as punctuated equilibrium.
Punctuated equilibrium has frequently been conflated with saltation theory, though the two theories are distinct. The difference is that the two theories describe punctuated phenotype change and punctuated genotype change respectively. Saltation theory suggests new species can be generated rapidly because of sudden large-scale mutation(s) – in other words the underlying genetic evolution causing the speciation event is itself punctuated (Figure 2B). Punctuated equilibrium on the other hand proposes that gross phenotypic change is the consequence of gradual (though perhaps rapid) genetic evolution in an isolated population. Richard Goldschmidt described the gross mutations as hopeful monsters – striving for ‘perfection’ in one big jump[58], however it is more likely that most gross genetic rearrangements will be maladaptive.
It is increasingly clear that the punctuated evolution of both phenotypes and genotypes occurs during cancer development.
Punctuated phenotype change is clearly seen in the development of neoplasia: typically, a neoplastic lesion (such as a colorectal adenoma)– with a grossly different phenotype to the normal cells – arises ‘abruptly’ without intermediate partial neoplastic forms (although we acknowledge that one could argue that crypt hyperplasia may sometimes be an intermediate form in the intestine). It is important to recognise that such punctuated
phenotype change (normal to malignant cells) may be underpinned by gradual genotype evolution. In the example of intestinal neoplasia, it is clear that loss of the normal function of the APC gene is sufficient to generate adenomas[30, 31]. Loss of normal APC function can be caused by the ‘gradual’ accumulation of the two mutational hits on each of the APC alleles[59], and is clear that the gross changes in phenotype (normal to neoplastic) need not be accompanied by large scale genetic alteration[60]. In leukaemia, ‘intermediate’ clone genotypes are present at only very low frequency, potentially indicating punctuated equilibrium-like evolutionary dynamics[61]. In follicular lymphoma, disease transformation is associated with an increased mutation burden and often also the acquisition of mutations in key ‘driver’ genes, though the underlying temporal pattern of mutation accumulation remains undetermined[21].
The genotype-phenotype map describes the relationship between genetic change and phenotypic traits. APC-loss in the intestine demonstrates how slight changes in genotype (eg single base-pair changes) can cause large changes in phenotype: this is an example where mapping between the space of possible genotypes and phenotypes is not smooth. Moreover, phenotypic change may not occur until multiple independent mutations in a number of key genes have accumulated and act in tandem to cause phenotypic alteration (this is called epistasis). Epistasis can underlie punctuated equilibrium in cancer – an individual lineage may steadily acquire individual driver mutations but not clonally expand until it has the full complement of drivers necessary to enhance its fitness. Intriguingly in colorectal[62, 63] and lung[20] cancers the majority of the driver mutations often appear clonal throughout the cancer, perhaps implying that the growth of these cancers could be initiated only when a complete epistatically-interacting complement of drivers are obtained. Epistasis clearly adds much complexity to the relationship between genotypes and phenotypes. Resolving the genotype-phenotype map is key to understanding evolutionary trajectories in cancer, though given the inherently contextual definition of phenotypes and the near infinite space of possible genotypes, the resolution will be extremely challenging to achieve.
It is increasingly clear that Goldschmidt’s hopeful monsters –punctuated changes in genotype – are frequently found in cancer (Figure 3). Chromothripsis – chromosome shattering and reassembly in an aberrant manner – has now been reported in many different cancer types[64-66] and this saltatory mutation occurs following a single ‘catastrophic’ mitosis[67]. Chromoplexy – the interleaving of different chromosomal regions into one aberrant block – has been reported in prostate cancer and is likely to occur in a single cell division[68]. More generally, genome doubling is a relatively common saltatory mutation type observed across cancer types[69], and furthermore the tolerance of genome doubling facilitates subsequent chromosomal instability[70]. In breast cancer, sequencing of individual nuclei detects clones with dramatic copy number deviation from the diploid genome and no evidence of cells with intermediate patterns of copy number alteration[71, 72] (Figure 3). Similarly relative homogenous intra-tumour patterns of grossly deviant copy number alterations are observed in many cancer types including colorectal[62] and ovarian[17] and the premalignant disease Barrett’s Oesophagus[25, 73] suggesting underlying saltatory mutational mechanisms.
How often are these hopeful monsters formed? The monsters we sample in cancer are the ones that have fortuitously stumbled upon an adaptive genotype-phenotype combination. But presumably many saltatory mutations lead to alters maladapted phenotypes, or phenotypes that are lethal – indeed this is often the case for chromosomal instability[74]. Logically therefore, this means that for every saltatory mutation that produces an adaptive phenotype, there are likely many more saltatory mutations that produces maladapted phenotypes. This likely abundance of ‘maladapted monsters’ in cancers types (or their premalignant precursors) that frequently show saltatory mutation is a testable prediction, and furthermore the detection of maladapted monsters could prove to be a useful prognostic biomarker in premalignant diseases such as Barrett’s Oesophagus where large- scale genome alteration appears to be a key punctuated event in cancer formation[25, 73].
An important aside is the potential for punctuated evolution of the rate of single nucleotide alterations (SNAs). SNAs accumulate according to a relatively small number of underlying mutational processes associated with natural replication errors, defective DNA replication and repair machinery and mutagen exposures[47]. Although the accumulation of SNAs is a clearly a gradual process (though as noted above, individual SNAs can cause punctuated change in phenotype), we note that the abrupt ‘switching on’ of a new mutational process can cause punctuated changes in the SNA mutation rate. For example, mutation of the mismatch repair machinery (MMR) causes a sudden increase in a cell’s point mutation rate[75].
Conclusion: neutrality and selection, and punctuation and gradualism are each two sides of the same coin
Cancer genomes reveal frequent evidence of both neutral evolution and clonal selection. Since neutral evolution is just the evolution that happens between selection events – e.g. the evolution that happens within a clone – the frequent detection of neutral evolution in cancer should come as no surprise. It is our opinion that in fact it would be more surprising if a signature of neutral evolution was never seen in cancer, because this would mean that new ‘driver’ mutations accrued all the time in our cells – an implication that appears at odds with the relatively low age-dependent incidence of cancer[76] and the small number of drivers with respect to passengers[40].
At a molecular level, cancers unquestionably show both gradual (the steady accumulation of single nucleotide variants) and punctuated (large scale copy number alterations) genotype change. But whether or not phenotype change is similarly punctuated depends upon the relationship between the genotype and phenotype, and also the microenvironment context. Thus, to be able to predict and manipulate the evolutionary trajectories of cancer for respective prognostic and therapeutic benefits, it is critical that we understand the genotype-phenotype map and the associated transitions around genotype-phenotype space. To achieve this, we critically need to understand exactly which phenotypes in cancer are selected and why – genetics help us to understand the accessibility of the space of different phenotypes, but alone genetics cannot give us the full picture of cancer evolution.
Finally, a word of caution. Studies show that treatment frequently selects for rare subclones in a tumour – and sometimes subclones that were so rare they went undetected in the pre- treatment samples[26, 77-80]. Thus, while the evolutionary dynamics of large tumour subclones – the focus of this review – are clearly of much interest to understanding of the basic biology of cancer evolution, we must ask ourselves if these dynamics directly relate to a patient’s prognosis. It is our conviction that these evolutionary dynamics are clinically relevant, since only by learning the ‘rules of cancer evolution’ can we hope to effectively intervene and change the evolutionary course.
Acknowledgements
TG and AS are grateful for support from the Wellcome Trust. TG is also a Cancer Research UK Career Development Fellow. AS is also supported by the Chris Rokos Fellowship in Evolution and Cancer. The funders had no influence on the content of this review. The authors are grateful to Marc Williams for providing images for Figure 2 and to Nicholas Navin for providing Figure 3.
Statement of author contributions
TG and AS co-wrote the manuscript.
References
- Alexandrov LB, Jones PH, Wedge DC, et Clock-like mutational processes in human somatic cells. Nat Genet 2015; 47: 1402-1407.
- Xue Y, Wang Q, Long Q, et al. Human Y Chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting Curr Biol 2009; 19: 1453-1457.
- Genomes Project C, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale Nature 2010; 467: 1061-1073.
- Laird CD, Pleasant ND, Clark AD, et al. Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA Proc Natnl Acad Sci U S A 2004; 101: 204-209.
- Lengauer C, Kinzler KW, Vogelstein Genetic instabilities in human cancers. Nature 1998; 396: 643-649.
- Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature 2004; 432: 338-
- Schwarz RF, Trinh A, Sipos B, et Phylogenetic quantification of intra- tumour heterogeneity. PLoS Comput Biol 2014; 10: e1003535.
- Shibata D, Tavare Counting divisions in a human somatic cell tree: how, what and why? Cell Cycle 2006; 5: 610-614.
- de Bruin EC, McGranahan N, Mitter R, et Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014; 346: 251-256.
- Gerlinger M, Horswell S, Larkin J, et Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46: 225-233.
- Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion N Engl J Med 2012; 366: 883-892.
- Thirlwell C, Will OCC, Domingo E, et al. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal Gastroenterology 2010; 138: 1441-1454, 1454.e1441-1447.
- Siegmund K, Marjoram P, Woo Y, et Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proc Natnl Acad Sci U S A 2009; 106: 4828-4833.
- Sottoriva A, Kang H, Ma Z, et A Big Bang model of human colorectal tumor growth. Nat Genet 2015; 47: 209-216.
- Jones S, Chen W-D, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor Proc Natnl Acad Sci U S A 2008; 105: 4283-4288.
- Yachida S, Jones S, Bozic I, et Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010; 467: 1114-1117.
- Schwarz RF, Ng CK, Cooke SL, et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: a phylogenetic PLoS Med 2015; 12: e1001789.
- Sottoriva A, Spiteri I, Piccirillo SG, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary Proc Natl Acad Sci U S A 2013; 110: 4009-4014.
- Sottoriva A, Spiteri I, Shibata D, et al. Single-molecule genomic data delineate patient-specific tumor profiles and cancer stem cell organization. Cancer Res 2013; 73: 41-49.
- Zhang J, Fujimoto J, Zhang J, et Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014; 346: 256-259.
- Okosun J, Bodor C, Wang J, et Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 2014; 46: 176-181.
- Nik-Zainal S, Van Loo P, Wedge DC, et The life history of 21 breast cancers.Cell 2012; 149: 994-1007.
- Yates LR, Gerstung M, Knappskog S, et Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 2015; 21: 751- 759.
- Gundem G, Van Loo P, Kremeyer B, et The evolutionary history of lethal metastatic prostate cancer. Nature 2015; 520: 353-357.
- Li X, Galipeau PC, Paulson TG, et Temporal and spatial evolution of somatic chromosomal alterations: a case-cohort study of Barrett’s esophagus. Cancer Prev Res (Phila) 2014; 7: 114-127.
- Diaz LA, Jr., Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012; 486: 537-540.
- Sieber OM, Tomlinson SR, Tomlinson Tissue, cell and stage specificity of (epi)mutations in cancers. Nat Rev Cancer 2005; 5: 649-655.
- Lipinski KA, Barber LJ, Davies MN, et al. Cancer evolution and the limits of predictability in precision cancer Trends Cancer 2016; 2: 49-63.
- Vermeulen L, Morrissey E, van der Heijden M, et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science 2013; 342: 995-
- Lamlum H, Papadopoulou A, Ilyas M, et al. APC mutations are sufficient for the growth of early colorectal Proc Natnl Acad Sci U S A 2000; 97: 2225-2228.
- Sansom OJ, Reed KR, Hayes AJ, et Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Devel 2004; 18: 1385- 1390.
- Aktipis CA, Boddy AM, Gatenby RA, et Life history trade-offs in cancer evolution. Nat Rev Cancer 2013; 13: 883-892.
- Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer 2008; 8: 415-424.
- Pepper JW, Sprouffske K, Maley Animal cell differentiation patterns suppress somatic evolution. PLoS Comput Biol 2007; 3: e250.
- Stratton MR, Campbell PJ, Futreal The cancer genome. Nature 2009; 458:719-724.
- Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome Science 2013; 339: 1546-1558.
- Roth A, Khattra J, Yap D, et PyClone: statistical inference of clonal population structure in cancer. Nat Methods 2014; 11: 396-398.
- Andor N, Harness JV, Muller S, et EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics 2014; 30: 50-60.
- Fischer A, Vazquez-Garcia I, Illingworth CJ, et al. High-definition reconstruction of clonal composition in Cell Rep 2014; 7: 1740-1752.
- Stratton MR, Campbell PJ, Futreal The cancer genome. Nature 2009; 458:719-724.
- Marusyk A, Tabassum DP, Altrock PM, et al. Non-cell-autonomous driving of tumour growth supports sub-clonal Nature 2014; 514: 54-58.
- Williams MJ, Werner B, Barnes CP, et Identification of neutral tumor evolution across cancer types. Nat Genet 2016; 48: 238-244.
- Bozic I, Gerold JM, Nowak MA. Quantifying clonal and subclonal passenger mutations in cancer PLoS Comput Biol 2016; 12: e1004731.
- Durrett Population genetics of neutral mutations in exponentially growing cancer cell populations. Ann Appl Probab 2013; 23: 230-250.
- Ostrow SL, Barshir R, DeGregori J, et Cancer evolution is associated with pervasive positive selection on globally expressed genes. PLoS Genet 2014; 10: e1004239.
- Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human Science 2015; 348: 880-886.
- Alexandrov LB, Nik-Zainal S, Wedge DC, et Signatures of mutational processes in human cancer. Nature 2013; 500: 415-421.
- Greenman C, Stephens P, Smith R, et Patterns of somatic mutation in human cancer genomes. Nature 2007; 446: 153-158.
- Driessens G, Beck B, Caauwe A, et Defining the mode of tumour growth by clonal analysis. Nature 2012; 488: 527-530.
- Merlo LMF, Pepper JW, Reid BJ, et Cancer as an evolutionary and ecological process. Nat Rev Cancer 2006; 6: 924-935.
- Beerenwinkel N, Antal T, Dingli D, et Genetic progression and the waiting time to cancer. PLoS Comput Biol 2007; 3: e225.
- Bozic I, Antal T, Ohtsuki H, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natnl Acad Sci U S A 2010; 107: 18545-18550.
- Cancer Genome Atlas Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330-337.
- Castro-Giner F, Ratcliffe P, Tomlinson The mini-driver model of polygenic cancer evolution. Nat Rev Cancer 2015; 15: 680-685.
- Andor N, Graham TA, Jansen M, et Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 2016; 22: 105-113.
- Morris LG, Riaz N, Desrichard A, et al. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of Oncotarget 2016; 7: 10051-10063.
- Eldredge N and Gould SJ. Punctuated Equilibria: an alternative to phyletic Chapter 5, pp 82–115. In Models in Paleobiology (ed. Schopf T), Freeman, Cooper & Co, 1972.
- Goldschmidt R. The Material Basis of Evolution. New Haven CT: Yale University Press,1940.
- Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: A mutational ‘hotspot’ and interdependence of the ‘two hits’. Proc Natnl Acad Sci U S A 2000; 97: 3352-3357.
- Jones A, Thirlwell C, Howarth K, et al. Analysis of copy number changes suggests chromosomal instability in a minority of large colorectal The Journal of Pathology 2007; 213: 249-256.
- Campbell PJ, Pleasance ED, Stephens PJ, et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep Proc Natl Acad Sci U S A 2008; 105: 13081-13086.
- Kim TM, Jung SH, An CH, et Subclonal genomic architectures of primary and metastatic colorectal cancer based on intratumoral genetic heterogeneity. Clin Cancer Res 2015; 21: 4461-4472.
- Jesinghaus M, Wolf T, Pfarr N, et al. Distinctive spatiotemporal stability of somatic mutations in metastasized microsatellite-stable colorectal Am J Surg Pathol 2015; 39: 1140-1147.
- Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer Cell 2011; 144: 27-40.
- Kloosterman WP, Hoogstraat M, Paling O, et Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol 2011; 12: R103.
- Molenaar JJ, Koster J, Zwijnenburg DA, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012; 483: 589-593.
- Zhang CZ, Spektor A, Cornils H, et Chromothripsis from DNA damage in micronuclei. Nature 2015; 522: 179-184.
- Baca SC, Prandi D, Lawrence MS, et Punctuated evolution of prostate cancer genomes. Cell 2013; 153: 666-677.
- Carter SL, Cibulskis K, Helman E, et Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012; 30: 413-421.
- Dewhurst SM, McGranahan N, Burrell RA, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome Cancer Discov 2014; 4: 175-185.
- Wang Y, Waters J, Leung ML, et Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014; 512: 155-160.
- Gao R, Davis A, McDonald TO, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast Nat Genet 2016; 48: 1119-1130.
- Stachler MD, Taylor-Weiner A, Peng S, et al. Paired exome analysis of Barrett’s esophagus and Nat Genet 2015; 47: 1047-1055.
- Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in Nat Rev Genet 2012; 13: 189-203.
- Tsao JL, Yatabe Y, Salovaara R, et al. Genetic reconstruction of individual colorectal tumor Proc Natnl Acad Sci U S A 2000; 97: 1236-1241.
- Armitage P, Doll The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer 1954; 8: 1-12.
- Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012; 481: 506-
- Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in Nature 2011; 469: 356-361.
- Findlay JM, Castro-Giner F, Makino S, et Differential clonal evolution in oesophageal cancers in response to neo-adjuvant chemotherapy. Nat Commun 2016; 7: 11111.
- Siravegna G, Mussolin B, Buscarino M, et Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015; 21: 827.
Figure Legends
Figure 1. The influence of selection in the cancer genome.
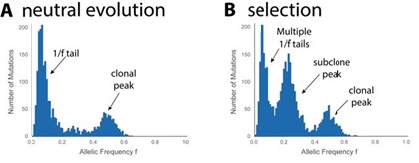
(A) A simulated distribution of variant allele frequencies (VAF) in a cancer that is evolving neutrally. These VAF
distributions are naturally produced by next generation genome sequencing. The distribution has a peak around 0.5 – these are the clonal variants present in all cancer cells. The distribution of subclonal variants (those at lower frequencies) follows a ‘1/f’ distribution whereby there are ever many more mutations at ever-lower frequency.
Neutral cancer evolution can be detected by comparing the observed distribution of mutation frequencies in a cancer to this expected theoretical distribution.
(B) VAF distribution for an in silico model of a cancer where a fitter subclone has clonally expanded within the tumour. The cluster of mutations within the subclone are ‘passenger’ mutations dragged along to higher frequency within the tumour during the clonal expansion. Even though there is selection, there is still a ‘background’ of neutral evolution – this is the neutral evolution within the selected subclone and in the residual tumour cells.
Figu e 2: Punctuated equilibrium and hopeful monsters.
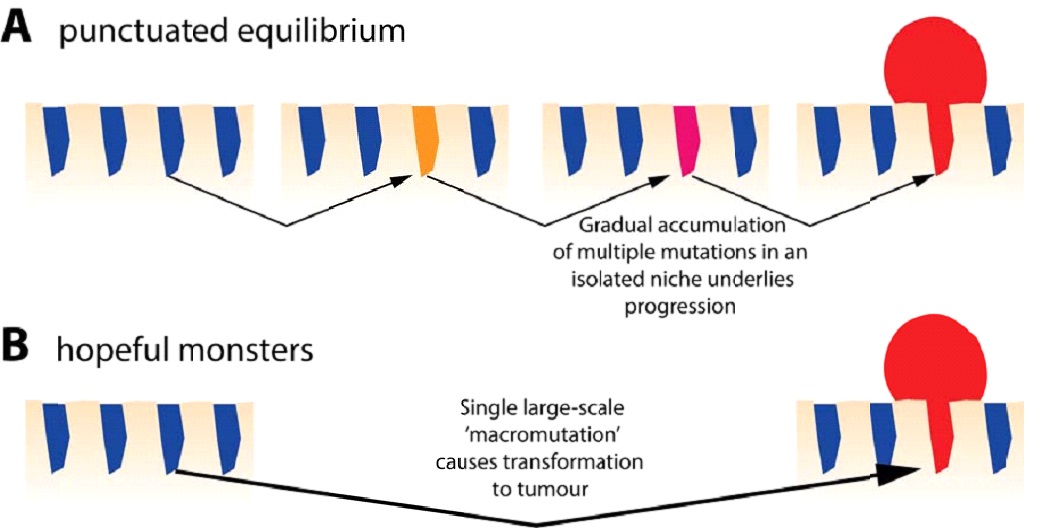
(A) Mutations accumulate within small spatially isolated niches (here an intestinal crypt is depicted) and only after a sufficient number of adaptive mutations are acquired is a clonal expansion initiated. From a macroscopic perspective, the evolution of the neoplasia appears punctuated, even though the driver mutations were acquired gradually within the crypt.
(B) The generation of a hopeful monster – a clone with a grossly altered genotype – in a single cell division produces a neoplastic in a single ‘catastrophic’ step.
Figure 3: Primary data indi ating punctuated copy number evolution.
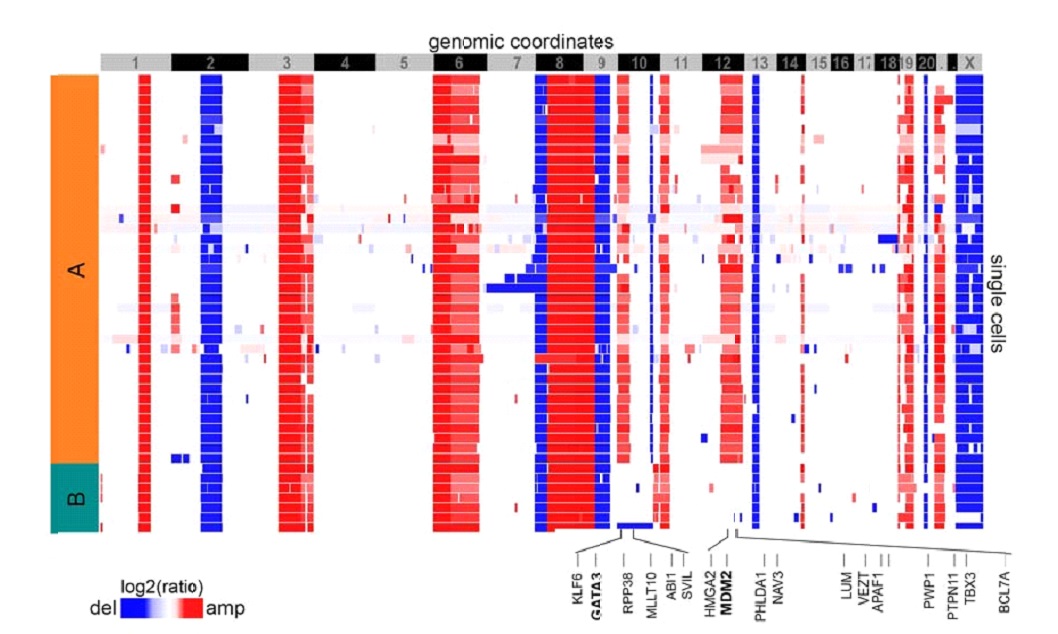
Copy-number profiles of individual breast cancer cells from a single breast cancer case showing the same grossly altered genomes are observed in all cells sampled, and no intermediate forms are detected. Image scaled, cropped and reprinted with permission from ref. [72].
Table 1. Definition of terms Neutral evolution
Evolution where all individuals in the population have equal fitness. In a growing population (like a newly formed tumour) this means that all cells grow at the same rate.
Drift
Stochastic effects (e.g. random cell death in a tumour) can cause some ‘lucky’ individuals in a population to have more offspring than another, and so the ‘lucky lineage’ increases in size. Consequently, drift can cause fluctuations in subclone size in the absence of selection.
Selection
The process that results in one individual in a population, because of its particular well- adapted traits, having more offspring than another less well-adapted individual.
Fitness
The relative ability of an individual to produce surviving offspring in a population.
Punctuated equilibrium
The process whereby apparently abrupt changes in phenotype of the population at large occur because of gradual evolution in small spatially isolated niches.
Hopeful monster/saltation
The process whereby abrupt changes in phenotype are cause by underlying (large scale) punctuated changes in the genome. In cancer, massive genome alterations occurring in a single cell division are examples of saltatory genome evolution.